SEMITIP V6, Technical Manual
Technical instructions are provided below for SEMITIP version 6, in the sections:
Additional information on the theory behind the SEMITIP package can be found in Refs. [1]-[6] and in the documents:
The SEMITIP version 6 software package contains a set of programs for computing the electrostatic potential and resulting tunnel current between a metallic tip and a semiconductor sample. An brief introduction to the package is provided at the main
SEMITIP V6 Web Page,
including a discussion of various dimensionalities, geometries, and types of tunnel current computation that can be handled. A different program is employed for each dimensionality, geometry, or type of current computation. Links to those programs, including documentation for each, are included on the main SEMITIP V6 Web Page.
The purpose of this Technical Manual is to describe
various methods employed in the algorithms, coding, or usage of the SEMITIP package that are common
to a range of the programs.
As mentioned above the SEMITIP package permits computations for many different dimensionalities, geometries, and types of tunnel current computations. The goal of the package is to both provide users with the capability to easily perform computations using existing programs suitable for their problem, and to enable them to develop new routines or programs for handling situations not covered by presently existing programs. This first goal could in principle be handled by a single master program that handled every possible type of situation. That code would, however, be very unwieldy since by its nature it would accomodate many different situations. For development of new code by users, generally there is some specific situation (i.e. dimensionality, geometry, type of current computation, etc.) that is being considered. Then, it is very much easier for a user to modify existing code that deals with that specific situation rather than dealing with code that covers every possible situation. This is the reason that separate programs have been developed to deal with the various dimensionalities, geometries, and types of tunnel current computation that are covered within the SEMITIP package.
All of the programs in the SEMITIP package take their input from a file named FORT.9 (i.e. with a different FORT.9 file for each program). Comments within each of those files specify the meaning of each parameter, with additional information for certain parameters provided below. The line numbers in
the description below are applicable to the
Uni2 program for parameters
having to do with computation of potential, to the
UniInt2 program for parameters having to do with computation of current by integration of the Schrödinger equation, to the
UniIntSC2 program for parameters having to do with self-consistency when computing the current by integration of the Schrödinger equation (only relevant for situations of inversion or accumulation), and to the
UniPlane3 program for parameters having to do with computing the current using a plane wave expansion. However, the relevant line numbers for the FORT.9 files of other programs can be easily deduced from those listed below since the parameters within every input file are clearly labeled within the file itself.
Parameters for computation of potential: (line numbers according to FORT.9 file of
Uni2)
-
line 1 - number of data sets - If the value of this parameter is set to 1, then the program just makes a single pass through the parameter set, reading in the values and executing the program accordingly. (Multiple voltages might be input, with the "number of voltage points" parameter at line 39, but other than that just a single pass through the functions of the program is made). However, if a user would like multiple executions of the program, e.g., multiple runs while varying some parameter such as tip-sample separation, then that can be accomplished by using a value >1 for this number of data sets parameter. With a value >1, all parameters in FORT.9 are input this specified number of times (except for the parameter value on line 1 itself, which is only input once), and the program executed accordingly.
-
line 2 - shank slope (dimensionless) = tan (90-(theta/2)) where theta is shank opening angle
-
line 3 - tip-sample separation (nm)
-
line 4 - tip radius (nm)
-
line 5 - radius of hemispherical protrusion at end of tip (nm) - the shape of a protrusion on the end of the tip is specified in the user-defined function P that occurs at the end of the main program (for further discussion see section on User Supplied Routines). By default, the protrusion has a hemispherical shape. It should be noted that the influence of such a protrusion is found generally to be small, so typically this parameter can just be set to zero.
-
line 6 - contact potential (work function of tip relative to sample) (eV) - see Eq. (6b) of Ref. [2], or equivalently Eq. (1b) of Ref. [3] or Eq. (2) of Ref. [4], for explicit definition of the contact potential (presently this parameter cannot be varied for differing areas of an inhomogeneous surface).
Lines 7-18 refer to parameters defining the semiconductor. For an inhomogeneous semiconductor containing various regions of different doping or various surface area with different surface charge densities, see section on Inhomogeneous Geometry.
-
line 7 - donor concentration (cm-3)
-
line 8 - acceptor concentration (cm-3)
-
line 9 - semiconductor band gap (eV)
-
line 10 - donor binding energy (eV)
-
line 11 - acceptor binding energy (eV)
-
line 12 - conduction band effective mass
-
line 13-15 - valence band effective masses (heavy hole, light hole, split-off hole bands) (in units of free electron mass) - SEMITIP performs computations for a valence band structure consisting of a 3 bands: light-hole, heavy-hole, and split-off. For the charge density, only the first two are used and they are combined into a "density-of-states" effective mass mdh=(mlh3/2+mhh3/2)2/3. It is this mdh that is used for computing charge densities in the valence band. For a computation of tunnel current, a separate computation is performed for each valence band, using its specified effective mass.
-
line 16 - spin-orbit splitting (eV) - this value is the energy of the split-off band below the other valence bands. This energy is not used in the computation of charge density (although it is still input into all programs, even if they don't need it), but it is used for the computation of tunnel current.
-
line 17 - semiconductor degeneracy indicator (=0 for nondegenerate, =1 for degenerate) - a degenerate semiconductor is one for which ALL of the electrons or holes introduced by doping occupy the respective conduction or valence band states. That is, they are never bound to the dopant cores, no matter how low the temperature is. (This is the correct definition of a degenerate semiconductor, as discussed e.g. in Ziman's Solid State textbook, even though many other Solid State textooks provide very poor definitions for this electronic phase).
-
line 18 - inversion indicator (1 or 2 to suppress VB or CB occupation, 3 for both, 0 otherwise) - as discussed in Ref. [2], depending on the size of the semiconductor band gap, inversion of the material might be impossible under the conditions of a typical STM experiment even of the band bending would suggest that inversion should occur. In other words, for large upwards band bending on n-type material, inversion would lead to occupation of the valence band states near the surface by holes. But, the current that flows across the bandgap in the semiconductor to supply those holes is highly dependent on the band gap magnitude. For band gaps greater than about 1 eV this inversion current in the semiconductor is very low, so that the tunnel current from the tip to the surface would immediately occupy those empty valence band states and consequently the inversion of the surface would not occur. Inversion has been observed for Ge (bandgap of about 0.7 eV), and the large gap of GaAs (1.4 eV) definitely precludes inversion from even occurring within an STM experiment. The situation for Si (gap of about 1.1 eV) is an intermediate one; based on the computations of Ref. [2] it seems unlikely that inversion would occur, but an experimental test of that is needed to definitely understand the situation. The parameter on line 18 allows the user to prevent inversion from occurring. To suppress the inversion, set the parameter to a value of 1 for n-type material (which prevents occupation of VB states of holes) or to 2 for p-type material (which prevents occupation of CB states by electrons). For undoped material a value of 3 can be used, to prevent occupation of both the VB by holes or the CB by electrons. Other settings for this parameter (e.g. a value of 2 for n-type material) leads to ill-defined, unphysical sitatutions and must be carefully avoided (the program does have some checks for these sorts of unphysical situations).
-
line 19 - dielectric constant of semiconductor (presently this parameter cannot be varied for differing regions of an inhomogeneous semiconductor).
-
line 20 - temperature (K)
Lines 21-28 refer to parameters that define the distribution in energy of surface states on the semiconductor. By default two types of distributions can be input, and the charge densities from these are summed together in the program for computing the total charge density (physically, many surface might have intrinsic and extrinsic surface states, the former from the states of the intrinsic surface atoms and the latter from defects on the surface, and hence the use of two distributions). The distributions themselves are defined in a user-defined function SIG which is part of the main program (for further discussion see section on
User-defined Functions).
The parameters below corresponding to the default form of the SIG function. By default two types of distributions are possible: a uniform distribution of states and a distribution containing two Gaussians. In the first case the parameters are just the density of states and the charge neutrality level [i.e. that separates states of acceptor character (negatively when occupied by an electron and neutral when empty) from those with donor character (neutral when occupied by an electron and positive when empty)]. To use this type of uniform distribution, then the value of the FWHM parameter should be zero (and the centroid energy is not used by the program, although it still should be present in the FORT.9 parameter list). For the Gaussian type distribution, nonzero values for the FWHM and centroid energies are supplied. The former is the full-width-at-half-maximum for each Gaussian and the latter is the position of the peak of each Gaussion relative to the charge neutrality level (there's one Gaussion below that level and one above it). One additional issue regarding surface states is whether or not the distributions change (e.g. go to zero) for energies with the bulk valence or conduction band. This constraint can applied by a line of the code within the SIG routine, and for the default versions of SIG there is no change of the distributions when the energies fall within the bulk bands.
-
line 21 - density of FIRST distribution of surface states (cm-2 eV-1 for uniform distribution; cm-2 for Gaussian distribution, corresponding to the energy-integrated value)
-
line 22 - charge neutrality level (eV), relative to valence band maximum
-
line 23 - FWHM for Gaussian distribution (use uniform distribution if zero) (eV)
-
line 24 - centroid energies for Gaussian distribution (eV)
-
line 25 - density of SECOND distribution of surface states (cm-2 eV-1 for uniform distribution; cm-2 for Gaussian distribution, corresponding to the energy-integrated value)
-
line 26 - charge neutrality level (eV), relative to valence band maximum
-
line 27 - FWHM for Gaussian distribution (use uniform distribution if zero) (eV)
-
line 28 - centroid energies for Gaussian distribution (eV)
-
line 29 - indicator for temperature dependence of surface state occupation (0=don't include it, 1=include it) - in situations where nonzero surface state densities are present, including the temperature dependence of the occupation of those densities adds significantly to the execution time for the program (i.e. when computing the table of surface charge densities). In nearly all cases this temperature dependence is not important, since at a given temperature the same effect produced by the temperature dependence can also be achieved by some slight variation in the parameters defining the surface states. Thus, setting this parameter to 0 causes the occupation of the surface states to be computed in the approximation of zero temperature.
Lines 30-36 refer to parameters that determine details of the finite-difference solution for the potential. This solution is performed first on an a specified number of grid points. Then, the number of grid points is doubled (and their spacing halved), and the computation performed on that doubled grid (using as an initial condition the solution from the previous grid). This procedure of doubling the number of grid points in each coordinate direction is referred to as "scaling" of the solution, and the scaling procedure is repeated a specified number of times. The finite-difference procedure itself is an iterative one, and within each scaling step it is performed until the change in Pot0 (the potential at the point on the semiconductor surface opposite the tip apex) is less than a specified convergence parameter value for both the present iteration and the previous one, or the number of iterations exceeds a specified maximum number. Concerning the initial spacing between grid points, this is straightforward for the points in the vacuum, but in the radial direction or in the direction into the semiconductor some estimation of the initial spacing must be made. This estimation of made based on values of semiconductor doping and tip radius as follows: First a 1D depletion length of the semiconductor is computed, sqrt(2 epsilon ΔV / e2 N ) where epsilon is the dielectric constant and ΔV corresponds to the tip-sample voltage or to 1 V, whichever is greater. The grid size is initially assigned the value of the tip radius. If the radius of a protrusion on the end of the tip is nonzero, then the grid size is assigned to that value or to its previous value, whichever is less. Then, the grid size is assigned to the above 1D estimate of depletion length divided by the number of grid points, or to its previous value, whichever is less. Finally, this value for the initial grid size is multiplied by the factor on line 33 (e.g. to reduce its value further, and hence achieve a finer grid). The grid size thus determined is used for both the radial direction and the z-direction into the semiconductor, except if the multiplier parameter on line 33 has a value <= 0. In that case, the following two additional lines provide the values of the grid size in the radial direction and the z-direction (or, for a 1D computation, just one additional line is used for providing the grid spacing in the z-direction). The resulting grid sizes are used to compute the spatially varying spacing between grid points using the algorithm described in the section on Spatial Coordinates.
-
line 30 - starting number of radial grid points
-
line 31 - starting number of grid points in the vacuum
-
line 32 - starting number of grid points in the semiconductor
-
line 33 - multiplicative parameter for initial grid size
-
line 34 - number of scaling steps for computation of potential
-
line 35 - maximum number of iterations in each scaling step
-
line 36 - convergence parameters for each scaling step
-
line 37 - size of table of charge densities - within SEMITIP a table of bulk charge densities is computed at the start of the program, in order to save computation time later during the finite-difference solution. The minimum and maximum energies in this table are estimated, and the size of the table is determined by this parameter on line 37. This size should be fairly large, around 20000 for high precision computations or 5000 for low precision computations.
-
line 38 - output parameter - this parameter determines the type and amount of output from the program, as specified under the detailed descriptions for each program. A value of 1 generally corresponds to basic output, 2 provides additional output, and 3 provides still more output (when wavefunctions are computed, higher values of this parameter are employed to control the output of the wavefunctions and corresponding charge densities).
-
line 39 - number of voltage points - this is the number of voltage values that will be input from the following line, with the program executed for each of these voltage values.
-
line 40 - list of voltage values (each value separated by a comma or a blank space)
-
line 41 - number of contours - this is the number of equipotential contours to produce for a contour plot of the potential (that plot is output for the output parameter >1, but the input values on lines 41 and 42 are required for any value of the output parameter).
-
line 42 - spacing of potential contours - if this value is 0, then the program uses a computed value according to number of contours and the minimum and maximum values of the potential.
Parameters for computation of tunnel current by integration of Schrödinger's equation: (line numbers according to FORT.9 file of UniInt2)
-
line 43 - electron affinity of semiconductor (eV)
-
line 44 - Fermi energy of tip (eV)
-
line 45 - number of parallel wavevectors for computation of current - value of 50 recommended for high precision computations, or 20 for low precision.
-
line 46 - number of energies for computation of current - value of 50 recommended for high precision computations, or 20 for low precision.
-
line 47 - target expansion factor in number of z values through the semiconductor and vacuum, for use when integrating Schrödinger's equation - the number of z values is chosen based on the maximum value of wavevector or inverse decay constant involved in the integration, multiplied by this expansion factor. That resulting value is then compared to the grid size, and a final expansion factor is determined. A parameter value of 100 is typically used for high precision computations, or 0 for low precision computations. When image potential is included (line 61), then this expansion factor is multipled by 10 for use in the vacuum region, since a very fine step size is required there.
-
line 48 - fraction of semiconductor depth to include in integration - this parameter is included as a time saver, since with the nonlinear step spacing employed for the solution to Poisson's equation, very large z values deep into the semiconductor are encountered. Such large z values are, however, not needed for the integration of Schrödinger's equation. This parameter specifies the fractional number of z values used in the solution to Poisson's equation that will be employed in the integration. Typically a fraction of 0.5 or 0.75 is quite sufficient.
-
line 49 - modulation voltage (V) - the modulation voltage (as in a lock-in amplifier) used in computing derivatives. The actual spacing of voltages employed in the computation is +/- sqrt(2) times this value.
-
line 50 - -s(V) ramp for V<0 (normally positive) (nm/V) - this parameter is employed to simulate a z-ramp in the spectroscopy, for the negative voltage side. If such a ramp is not used, just set the parameter to zero.
-
line 51 - s(V) ramp for V>0 (normally positive) (nm/V) - this parameter is employed to simulate a z-ramp in the spectroscopy, for the positive voltage side. If such a ramp is not used, just set the parameter to zero.
-
line 52 - starting voltage of spectrum (V) - used in conjunction with the values on lines 50 and 51. (If those values are zero, then the value of this line 52 parameter is irrelevant).
-
line 53 - indicator for image potential (0=don't include it, 1=include it) - when this parameter is set to 1, an image potential is included in the potential within the vacuum region, as described in
Computation of Tunnel Current for Bulk States (including consideration of image potential and field-emission resonances).
Bulk charge densities are computed in the effective-mass approximation, using equations as specified in Ref. 1. These charge densities are evaluated repeatedly within the programs - at each grid point, each iteration of the solution, and each 1D search step to solve the nonlinear aspect of Poisson's equation. To reduce the computation time needed for these evaluations, a table of charge density values to computed at the start of each program (or multiple tables are computed, for an inhomogeneous semiconductor). The maximum possible number of elements in this table is specified by NEDIM in the PARAMETER statement within each program, and the actual number of element used is specified in the FORT.9 input file (line 37). The program uses a particular algorithm to decide on the energy bounds of this this table, and if a charge densities value is needed that falls outside of these bounds then that charge density is evaluated from the defining equations.
The presence of electronic surface states give rise to surface charge densities. There are in general two broad categories of surface states that can exist on the semiconductor, one being intrinsic states from the dangling bonds that exist within each unit cell of the structure, and the second being extinsic states that are sparsely distributed over the surface and arise from defects such as adsorbates or surface steps. For surfaces such as GaAs(110) that do not have any intrinsic states within the band gap, tip-induced band bending plays a particularly large role. Such surface can still have extrinsic surface states, which will affect this band bending. (Also, even for GaAs(110), intrinsic surface states located within the conduction band play an important role for situations of electron accumulation, as discussed in Ref. [5]). By default, the SEMITIP programs allow for input of two type of surface charge densities. Both of these densities are treated identically within the program, with the reason for allowing two inputs being to treat situations such as GaAs(110) for which both intrinsic and extrinsic states can play a significant role in determining the band bending.
Surface charge densities are defined by user-specified distribution(s), defined in the routine SIG. In the default form of those routines, a distribution that is uniform in energy, or one consisting of two Gaussian functions, are provided, but any other function can also be specified. (In principle, a function that varies very quickly with energy may be difficult for the program to handle, but no such limitation has yet been encountered). In addition to the energy distribution, the charge neutrality level for the distribution must also be specified, which is the energy above which the states are negative when filled and neutral when empty (i.e. acceptor like, as for conduction band states) and below which they are neutral when filled and positive when empty (i.e. donor like, as for valence band states). In analogy to the procedure for bulk charge densities, a table of surface charge densities is constructed at the start of each program. For the surface charge densities in particular this table assumes a zero temperature form for their occupation. Computations using occupations computed at nonzero temperature are possible (using the switch on line 29 of the FORT.9 input file), but in that case the table is not used and the occupations are always computed from the defining equations, thus requiring considerably more computation time. It is rare that the explicit inclusion of the temperature dependence of the occupation significantly affects the results, i.e. to an extent beyond what could be accomplished by a slight shift in one of the surface state parameters (such as the charge neutrality level), so that in most cases this temperature dependence need not be included in the computation.
It is important to realize that SEMITIP, in its present form, does not allow for any computations of tunnel current associated with the defined distributions of surface states. Rather, the definied charge density of the surface states is used only in computing the electrostatic potential, whereas the tunnel current is based entirely on bulk bands (possibly including localized regions of the potential as occur e.g. for a buried quantum dot). In some future version of SEMITIP it is possible that currents due to surface states could be included; the surface states in that case would be defined by a surface band structure, and the charge density for that band of states would be computed by an integral of the band structure (rather than be a user-defined density-of-states, as presently used).
A computation of electrostatic potential is said to be self-consistent if the solution for the potential contains full and proper dependence on the electronic states of the problem, with the electronic states themselves depending on the potential. The default mode of charge density computation within SEMITIP is a semi-classical one, in which defined bands of bulk or surface states are shifted in accordance with the electrostatic potential and they are occuppied according to a fixed (known) Fermi energy.
For a semiconductor in depletion, this type of computation is automatically (trivially) self-consistent, since the only relevant charge density of the problem is that due to the ionized dopants and this charge density is accurately treated in the semi-classical approximation.
Incorporating user-defined distributions of surface states into the solution for the potential is also self-consistent in the same trivial sense (assuming that those surface states are not modified by the application of the potential).
Nontrivial situations of self-consistency occur for accumulation or inversion in the semiconductor. These situations are not properly handled by a semi-classical treatment (at least not when only one or a few quantum states are occupied), but the
UniIntSC1
or
UniIntSC2
programs are specially designed to treat these cases. The quantum states are computed by the intcurr
routine, i.e. the same routine that is used for computing tunneling currents, but now used for constructing charge densities are needed for the self-consistent computation. The computation of the 3D charge densities is performed as a series of 1D computations at different r values, something that is appropriate only for a potential that varies slowly in the radial direction.
For self-consistent computations with an even more localized potential, e.g. as might occur at a quantum dot or near a point charge on a surface, the computation of the charge densities would have to employ the plane wave expansion method, as in
UniPlane3.
The result would be limited to a limited region about the point on the surface opposite the tip apex, i.e. a periodic repetition of this region. In any case, a program for handling such computations has not been developed to date.
A problem of considerable interest is the computation of the potential (and possibly the current) in the presence of a fixed charge, e.g. a point charge, on or near a surface.
SEMITIP was not originally designed to handle fixed charges; rather, the parameters within the FORT.9 file refer everywhere to charge densities that vary in accordance with the Fermi energy. Fixed charges, whose magnitude does not vary with the Fermi energy, can still be handled in the program, but they are handled in a slightly "kludgy" manner making user-defined modifications to the RHOSURF or RHOBULK routines. See
example 6 of Uni2 or
example 2 of Uni3. In these examples, the location and magnitude of the fixed charge
is explicitly defined with the RHOSURF or RHOBULK routine. An alternative method, of
course, is for the user to modify the input stream of the FORT.9 file to include such parameters, and they then would be passed to the RHOBULK or RHOSURF routines through a user-defined COMMON block.
It is important to note that a number of limitations exist for computations involving fixed charges:
-
First, the reader is reminded that the default manner in which SEMITIP handles all charge densities is a semi-classical one. This method is only valid for a slowly varying potential. Some situations that require explicit quantum computations are also handled within SEMITIP, such as semiconductor accumulation or inversion, but the package does not automatically handle all such situations. For a point charge on or near a surface, the electrostatic potential when the semiconductor is in depletion can be handled by the program (subject to some additional limitations described below), but situations of accumulation around the point charge (that is, occupation of localized states arising from the point charge as well as screening by extended states around the point charge) are not properly handled by the present package since it does not handle self-consistent computations with a plane wave basis. (Even if it did handle such computations, the limited spatial extent of any plane wave type computation would also be a significant restriction on the results).
-
A second limitation in defining fixed charge on the surface or in the bulk has to do with the grid used in the finite-difference computation. As illustrated in
example 6 of Uni2 and
example 2 of Uni3,
the spatial extent of the charge is defined by statements within the RHOSURF or RHOBULK routines. However, if that spatial extent does not precisely overlap with the areas or volumes subtended by the grid points of the computation, then the actual charge that is computed will differ from the intended charge. Hence, a sufficiently fine grid and/or an extended charge must be used so that this discrepency is not too large. For a charge located some distance from the tip position (central axis), it's no problem to model that charge as being extended over some region (since that effect of the charge is essentially the same as if it were localized at a single point). But, as the charge approaches the central axis, then this area or region over which it is definied must become smaller and smaller, with the grid size for the computation necessarily being correspondingly small. The user must ensure that these sizes for the spatial extent of a fixed charge and for the grid are appropriate to the particular situation being considered.
Poisson's equation, including the boundary condition at the semiconductor surface and possible nonlinearity arising from bulk or surface charge densities, is solved according to the method described in Ref. [1] (also see footnote 36 of Ref. [2] and point 1 of the
Internal Parameters section below).
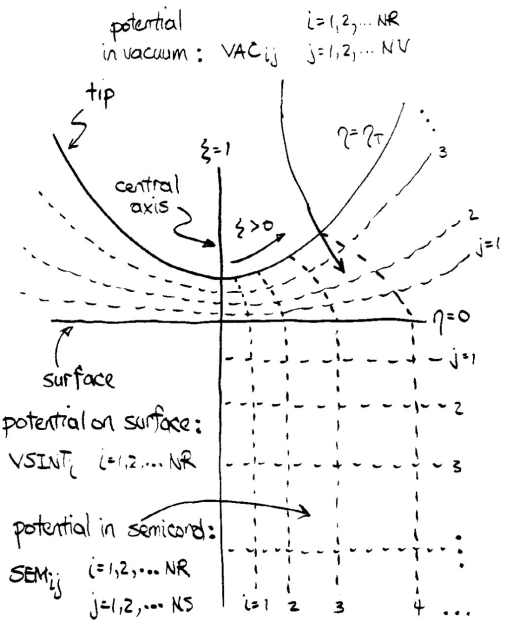
The solution for the potential is accomplished using cylindrical coordinate in the semiconductor and generalized prolate spheroidal coordinates in the vacuum. The latter are chosen to precisely match the shape of the probe tip (not including any protrusion on the end of the tip), as described in Ref. [4] and in Coordinate System
with accompanying
Diagram.
The spacing of the grid points in these coordinates is not uniform, but it increases as the radial distance r away from the central axis increases or as the distance z into the semiconductor increases. For computation of the tunnel current, this type of grid with quite large step sizes at large r or z is not suitable, and hence new grids with more nearly uniform spacing are formed. This process is done by the routine POTEXPAND for a tunnel current computation by integration of the Schrödinger equation or by POTPERIOD3 for a computation using the plane wave expansion method).
SEMITIP presently does not allow computation of tunnel currents from surface states. For the case of bulk states, two methods are available for computation of the tunnel current. In both, the Bardeen method together with the Tersoff-Hamann approximation for a sharp tip is used to write the current as a summation of the quantum states of the semiconductor (and the metallic probe tip). To obtain those states, the first method employs a "central axis approximation" in which numerical integration of the Schrödinger equation is performed only along the central axis of the problem. This method would be an exact solution for a planar geometry; it is an approximation for a nonplanar geometry but it still works quite well even for probe tips as sharp as 1 nm radius-of-curvature (see Ref. [6]). However, if regions of localized potential exist in the semiconductor, such as those due to a quantum dot, then that method in not applicable. Hence, the second available method is an expansion using plane waves (matched to decaying exponential in the vacuum), from which the solution of the Schrödinger equation is found by the usual eigenvalue method. Due to the significant computational demands of this plane wave method, it can be applied only over a limited region of space centered around the point on the semiconductor surface opposite the tip apex.
A new feature in version 6 of SEMITIP is the ability to handle inhomogeneous semiconductors, with the inhomogeneity existing in the bulk or on the surface. Actually, even in the prior versions of SEMITIP it was possible to handle certain special types of inhomogeneity (such as the presence of fixed charges) by explicit modification of the RHOBULK or RHOSURF routines, as described above under
Specification of Fixed Charge on Surface or in Bulk.
But within version 6 a general method for handling inhomogeneity in many of the parameters describing the bulk or surface has been implemented.
Such inhomogeneity does not represent any problem within a finite-difference type of computation that SEMITIP employs for the electrostatic potential, although whether or not a computation of tunnel current can truly handle an inhomogeneity depends very much on the range of the varying potential, as further discussed under
Computation of Tunnel Current.
Inhomogeneities within the bulk of the semiconductor are referred to as different "regions" of the material, and inhomogeneities on the surface are referred to as different "areas". The user defines the spatial boundaries of the different regions or areas by using user-defined functions RHOBULK or RHOSURF, respectively, as described in the following section. Parameters specifying the properties of the different regions or areas (e.g. their doping or their surface state density) can, in most cases, be input in the normal fashion from the FORT.9 input file.
The
Mult1,
Mult2, and
Mult3 programs
give examples of these procedures.
Again, for specification of fixed charges within the bulk or on the surface the situation is different, as described under
Specification of Fixed Charge on Surface or in Bulk.
In the present form of SEMITIP V6, computations of tunnel current for an inhomogeneous geometry are made using only a constant effective mass in the semiconductor. That mass is the one from the first defined region of the semiconductor. Thus, even if different masses for the various regions are defined within the FORT.9 input file, only the effective mass from region 1 is used in the computation of the current. (This limitation could be removed with some modest extension of the intcurr routine, but that extension has not yet been implemented).
When multiple regions are defined in the bulk material, then the locations of their respective valence band maxima can differ. Many of the quantities intput or output from SEMITIP (e.g. Fermi energy, charge neutrality level, etc.) use the valence band maximum as a zero of energy. In the situation with multiple regions, it is the valence band maximum of the region located at the origin of the coordinate system that is used as a zero of energy for all quantities, i.e. even those quantities associated with other regions.
A variety of user-defined functions are included in the same file as the main calling program. These functions allow definition of functions which cannot, in general, be specified simply by numbers in the FORT.9 input file. For example, a protrusion of arbitrary shape can be appended onto the end of the hyperbolic tip. Or, surface states of any arbitrary distribution in energy can be defined and their effect included in the computations of the potential. All user-defined functions are already included in the general purpose programs of the SEMITIP package, with default values for the functions as specified below:
-
An auxiliary function that defines a protrusion on the end of the probe tip is specified by a function P. The default shape for the protrusion is a hemisphere, but if some other function is desired then it can be defined by editing this routine and recompiling it. (For most computations a protrusion is not needed at all, so just using the default hemispherical protrusion with zero radius is fine for most cases). It should also be noted that the finite-difference grids needed to accomodate a nonzero protrusion can have relatively small spacing, hence requiring large numbers of grid points in order to extend far enough out in space for the potential to truly reach zero. Convergence in these cases will be problematic. Hence, considerably care must be applied if a nonzero protrusion is used, particularly if the doping employed for the semiconductor is low.
-
The energy distribution of surface states is specified in function called SIG. The default distributions are either uniform or Gaussian in energy. Different distributions can be achieved by editing this routine and recompiling it (see
example 3 of Mult3).
.
-
The spatial arrangement of both surface charge density and bulk charge densities are specified in functions called RHOSURF and RHOBULK. For example, surface states might be located only at particular surface regions as would occur for steps on a surface, or bulk doping might vary in the semiconductor as would occur for a pn junction. By default the surface states and bulk states are taken to be spatially uniform. Spatial nonuniformity can be achieved by editing these routines and recompiling them. In addition to defining possible spatial arrangements of charge, the RHOSURF and RHOBULK routines also check to see if the charge density for a given Fermi energy is available in the tables of charge densities. If so then that value is returned, or if not then the required charge density value is computed. Additionally, for self-consistent computations, these functions supply not just a semi-classical charge density but a fully self-consisent one using additional information passed from the main program through a COMMON block named CD (see discussion of COMMON blocks below).
-
The spatial arrangement of valence and conduction band edges are specified in functions called VBEDGE and CBEDGE. For both homogeneous and inhomogeneous semiconductors there is likely no reason to change the form of these routines from their default declarations. However, for the inhomogeneous case, the spatial information on the regions in the semiconductor is passed to VBEDGE and CBEDGE from another function called IGETREG, and this function could of course be change to accomodate different regions in the semiconductor (this auxiliary function obviates the need for multiple definitions of the different regions, in VBEDGE and CBEDGE separately). This same routine IGETREG could also be used in RHOBULK for identifying regions. It should be noted that the usage of the VBEDGE and CBEDGE routines is slightly different in the xxxInt programs employing integration of the Schrödinger equation compared to the xxxPlane programs employing the plane wave expansion. In the former, VBEDGE and CBEDGE provide the total band edge energy, whereas in the latter they provide only the change in energy relative to the band gap specified in the FORT.9 input file.
The vast majority of SEMITIP is written using standard FORTRAN features, and it thus should be compatible with any FORTRAN platform. The programs are however designed for use under Gnu FORTRAN, and possibly a few compiler-specific FORTRAN features may be employed. For example:
-
Use of PARAMETER statements in defining array sizes.
-
Frequent use of IF, THEN and IF, THEN, ELSE statements.
-
Input and output is directed to a specified logical unit number (LUN). Denoting this LUN by xx, then input comes from the file FORT.xx and output goes to FORT.xx.
-
Under Gnu FORTRAN, the default size of both integers and real numbers is 32 bits (4 bytes). This size is reflected in the record length for the binary WRITE statement of the entire potential to FORT.13. This record length (and possibly the form of the associated OPEN statement) may have to be changed for a different compiler.
COMMON blocks serve an important role in the SEMITIP programs by enabling the passing of parameters between the main programs and the user-defined functions, and in some cases to various supporting routines as well. The declarations and role of the various COMMON blocks is specified below. In situations when the block is only used between the main program and a user-defined function (i.e. it does not appear in any supporting routine), then modification by the user to accomodate some new situation is possible.
- Parameters defining the semiconductor are passed between the main program and many of the supporting routines as a COMMON block. For a homogeneous semiconductor this declaration appears as
COMMON/SEMI/TK,EGAP,ED,EA,ACB,AVB,CD,CA,IDEG,IINV
where the definition of the variables can be found in the source code of each main program. For an inhomogeneous the declaration is
COMMON/SEMI/TK,EGAP(NREGDIM),ED(NREGDIM),EA(NREGDIM),ACB(NREGDIM),
&AVB(NREGDIM),CD(NREGDIM),CA(NREGDIM),IDEG(NREGDIM),IINV(NREGDIM),
&DELVB(NREGDIM)
where NREGDIM is the maximum possible number of different types of regions in the semiconductor. For an inhomogeneous semiconductor this COMMON blocks also serve to pass information between the main program and the user-defined functions VBEDGE and CBEDGE that define the energies of the band edges.
-
Information concerning a possible protrusion on the apex of the probe tip is passed between the main program and the user-defined function P in a COMMON block declaration appearing as
COMMON/PROTRU/RAD2
where RAD2 is the radius of the hemispherical protrusion. This block occurs only in the main program and in the function P, so modification of the block (e.g. to accomodate some other type of protrusion) is possible.
-
Information on the distributions of surface states is passed between the main program and the SIG routine using a COMMON block which for a homogeneous semiconductor appears as
COMMON/SURF/ISTK,TK,EN0,EN(2),DENS(2),FWHM(2),ECENT(2)
with the dimension "2" referring to the possible use of two distributions of states (e.g. one intrinsic and one extrinsic). For an inhomogeneous semiconductor the declaration is
COMMON/SURF/ISTK,TK1,EN0(NARDIM),EN(NARDIM,2),DENS(NARDIM,2),
&FWHM(NARDIM,2),ECENT(NARDIM,2)
where NARDIM is the maximum possible number of different types of areas on the surface.
These blocks occur only in the main program and the SIG routines, so modification (e.g. to accomodate more parameters for some different distribution of surface states) is possible.
-
Charge densities of both surface and bulk states are passed in a COMMON block which for a homogeneous semiconductor appears as
COMMON/CD/EF,ESTART,DELE,NE,RHOBTAB(NEDIM),RHOSTAB(NEDIM)
where NEDIM is the dimension for the tables of charge densities.
For an inhomogeneous semiconductor the declaration is
COMMON/CD/EF,ESTART,DELE,NE,RHOBTAB(NREGDIM,NEDIM),
&RHOSTAB(NARDIM,NEDIM)
These COMMON blocks are used to pass information between the main program and the user-defined functions RHOBULK and RHOSURF as well as to various supporting routines.
Nearly all the parameters that control the functioning of the SEMITIP programs can be accessed through either the FORT.9 input file, the user-defined functions, or the PARAMETER and COMMON statements described above. There are however a few additional parameters that are internal to certain routines, modification of which require editing the source code and then re-compiling and linking. These internal parameters include:
-
A one-dimensional search is used as part of the finite-difference method for solving the nonlinear Poisson equation, as described in Ref. [1]. This search is performed to an accuracy in terms of energy of max(PotTip,1)/106, where max(PotTip,1) is the potential on the probe tip or 1 eV, whichever is greater. This precision parameter occurs several times within the semitip1, semitip2, and semitip3 routines. It is difficult to imagine a situation where this parameter would need to be changed, since a worse precision would not save a significant amount of computation time and it's unlikely that a higher precision would ever be needed (except for situations with very low temperature and a small energy scale, in which case possibly the precision of the entire computation would also require modification).
-
The IBC parameter specifying the boundary condition for the potential computation is set to 0 (Dirichelet boundary condition) at the top of the semitip1, semitip2, and semitip3 routines. See
Boundary Conditions
for more discussion of this parameter. As discussed there, a value of 1 for Von Neumann boundary conditions can be used, but it's useful only for particular cases and the value of 0 is most suitable for general purposes.
-
For both bulk and surface charge densities, tables of those values are constructed in the main program as discussed in the sections on
Computation of Bulk Charge Densities
and
Computation of Surface Charge Densities.
The energy bounds of those tables is determined by a particular algorithm within the main program. For some special situations charge densities might be required that are outside this range, although that's not a problem since in those cases the densities are evaluated from the defining equations. Nevertheless, if it is desired for some reason to change the bounds of the charge density tables, than that must be done by modifying the source code in the main program.
-
For self-consistent computations, there's a parameter called INITCD within the UniIntSC1 and UniIntSC2 programs that controls the initialization of the array used for constructing the charge densities. By default this parameter to set to 1, which corresponds to the array being initialized to semi-classical values. An alternative is INITCD=0 which initializes the array to zero, but that hasn't proved useful in any computations to date.
-
For computation of tunnel current, a two-band model for the band structure is not used. There are situation however where use of this model is appropriate, as discussed in
Kane's Two-band Model.
That document also explains how to modify the intcurr routine to enable a two-band type computation.
Due to the considerable expansion in the capability of SEMITIP version 6 compared to versions 1-5, a number of changes to the naming conventions for programs and variables were needed. These changes included:
-
In versions 1-5, numbers 1, 2, ... appended to the end of a routine (or program) name generally referred to different versions (or types) of the particular routine. In contrast, for version 6, numbers immediately following a routine name refer to the dimensionality that that routine applies to, whereas the version of the routine is given following a dash at the very end of the name. For example, SEMITIP_V5 referred to version 5 which computed the potential and the current (using integration along the central axis), self-consistently. In version 6, this becomes UniIntSC2-6.3, with "Uni" referring to a uniform semiconductor, "Int" referring to solving Schrödinger's equation by integration in the central axis approximation, "SC" referring to a self-consistent version of the code employing charge densities computed quantum mechanically, and the version being 6.3 (this follows the final version of SEMITIP_V5 which was 5.2). As another example, the routines potcut1.f and potcut2.f were used in versions 4 and 5 for obtaining a cut of the potential from the central axis, and expanding the grid of that cut, respectively. In the new versions, the former of these routines became potcutX_6.0.f with X=2 or 3 corresponding to the dimensionality, and the latter was renamed to potexpand_6.0 (no dimensionality indicator is needed). Additionally, the main finite-difference routine in the old version was named semistip.f (the middle "s" referring to inclusion of surface states), but in the new versions this is just called semitip.f. A listing of all available programs and routines in version 6 is found on the main
SEMITIP V6 Web Page.
-
Some variables in the programs have changed names between versions 1-5 and version 6. In particular, the term PHI was used to refer to electrostatic potential energy in versions 1-5, but in the version 6 this term is used for the azimuthal angle. Hence, PHI in the versions 1-5 becomes Pot in version 6, PANGLE becomes Phi, and DELPHI (for the contact potential) becomes CPot.
Additionally, in version 6, considerable repackaging of the routines occurred so as to enable the common usage of particular routines across many of the specific calling programs. The new program structure is described in the sections above.
For users upgrading from versions 4 or 5 to version 6, a few small changes in the input files (FORT.9) should be noted. In version 6, all input related to the computation and plotting of the potential is given before that for the computation of current. Hence, e.g. whereas in versions 4 or 5, specification of parameters for the contour plots occurred at the very end of the input file, those entries now occur at the end of the entries for the potential computation, and they are followed by the input parameters (if any) for the computation of the tunnel current.
One technical change occurred in version 6, namely, the introduction of a new method for computing derivatives for the variable-spacing grids that are employed in the semitip.f finite-difference routine. This method is further described in
Computation of Derivatives for Variable Grid Spacing.
References
1. R. M. Feenstra, Electrostatic Potential for a Hyperbolic Probe Tip near a Semiconductor, J. Vac. Sci. Technol. B 21, 2080 (2003). For preprint, see
http://www.cmu.edu/physics/stm/publ/52/.
2. R. M. Feenstra, S. Gaan, G. Meyer, and K. H. Rieder, Low-temperature tunneling spectroscopy of Ge(111)c(2x8) surfaces, Phys. Rev. B 71, 125316 (2005). For preprint, see
http://www.cmu.edu/physics/stm/publ/65/.
3. R. M. Feenstra, Y. Dong, M. P. Semtsiv, and W. T. Masselink, Influence of Tip-induced Band Bending on Tunneling Spectra of Semiconductor Surfaces, Nanotechnology 18, 044015 (2007). For preprint, see
http://www.cmu.edu/physics/stm/publ/74/.
4. Y. Dong, R. M. Feenstra, M. P. Semtsiv and W. T. Masselink, Band Offsets of InGaP/GaAs Heterojunctions by Scanning Tunneling Spectroscopy, J. Appl. Phys. 103, 073704 (2008).
For preprint, see
http://www.cmu.edu/physics/stm/publ/79/.
5. N. Ishida, K. Sueoka, and R. M. Feenstra, Influence of surface states on tunneling spectra of n-type GaAs(110) surfaces, Phys. Rev. B 80 075320 (2009).
For reprint, see
http://www.cmu.edu/physics/stm/publ/85/.
6. S. Gaan, G. He, R. M. Feenstra, J. Walker, and E. Towe, Size, shape, composition, and electronic properties of InAs/GaAs quantum dots by scanning tunneling microscopy and spectroscopy, J. Appl. Phys. 108, 114315 (2010). For preprint, see
http://www.cmu.edu/physics/stm/publ/93/.
{kind=link}